Start for Beginner to Learn Computational Drug Design, Molecular Docking, Computer Aided Drug Design, Molecular Dynamics
What you will learn
Drug Retrieval
Single Software used for docking
Prediction to inhibit Viral Protein
Compound used as Drug Agent
Molecule Operating Environment (MOE)
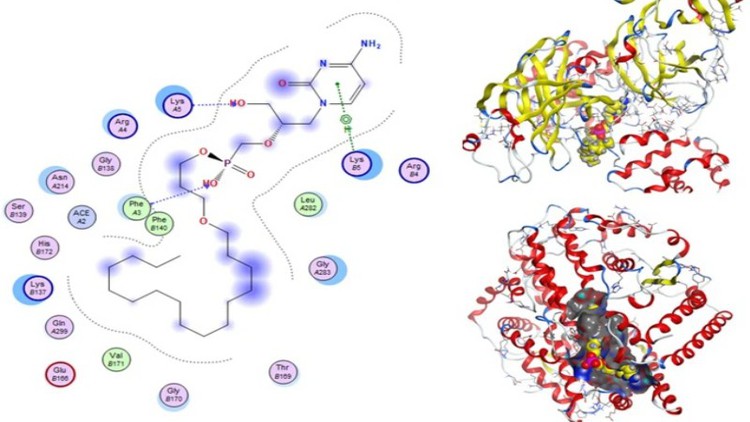
Ligand and Protein molecules interaction
Visualization 2D&3D Molecules interaction
How to generate publication quality figures from the docking output
Description
In the filed molecular orientation modeling, Molecular Docking the perfect binding of two molecules, like prediction of ligand binding on the active size of the protein. On the basic knowledge of computer you learn ligand based Computer Aided Drug design (CADD) approach involves the analysis of ligands known to interact with a target of interest. One such course is particularly designed to maintain knowledge at the beginner level of computer Drug Discovery applications for science students. Most easily Docking Software than the AutoDock. This short course will help students get a good start in becoming proficient in the field of docking and drug development simulation studies before they become familiar with the use of MOE software and dive into lab validation studies. A real problem of today’s world was taken as an example in this course and a drug agent called lutein which is present in papaya for quad “protein resistance and possibly drug agent capabilities. Was tested.
By the use of this software, we have performed the molecular docking studies of various naturally occurring compounds, anti virus, anti fungals, anti-nematodes and anti-protozoal drug by the pharmaceutical industry.
Throughout this course, you will discover Molecular Docking from scratch, including
- Install Molecular Docking Environment (MOE)
- Retire Ligand from Bioinformatics Database
- Get Protein sequence form Protein Data Bank (PDB)
- Performed Molecular Docking
- 2D & 3D Molecules Interaction
Content